| 対象疾患 | 治療ライン | 研究の相 | 評価項目 | 実施地域 | 日本の参加 |
|---|---|---|---|---|---|
| 進行消化管間質腫瘍(GIST) | 二次治療 | 第3相 | 腫瘍増悪までの期間 (中央判定) | 国際 | なし |
試験名 :なし
レジメン:スニチニブ vs プラセボ
登録期間:2003年12月〜2005年1月
背景
消化管間質腫瘍(GIST)は肉腫の一種であり、消化管で最も一般的な間葉系腫瘍である。大部分のGISTは膜貫通型受容体チロシンキナーゼであるKITを発現しており、これらの腫瘍のサブセットは明らかに悪性度が高く、40%以上で転移性病変を有すると考えられている。GISTの約85-90%はKITキナーゼの活性化につながるKIT 遺伝子の機能獲得型変異を有し、5%はPDGFRαをコードするPDGFRA 遺伝子の機能獲得型変異を有しており、KIT とPDGFRA 遺伝子の活性型変異は大部分のGIST症例において悪性表現型の発生と維持の背景に存在している。このような分子病態生理を理解することにより、シグナル伝達異常を標的とする薬剤開発が可能となった。
KITおよびPDGFRのキナーゼ活性を選択的に阻害するイマチニブは進行GISTの有効性を大幅に改善した。しかし進行GISTの5%はイマチニブに対する一次耐性を、また14%は早期耐性を示し、獲得耐性は治療開始後約2年(中央値)に出現する。しかし、イマチニブ不応となったGISTに対する有効な治療薬はなく、新たな治療薬の開発はアンメットメディカルニーズであった。
スニチニブはin vitroおよびin vivo腫瘍モデルで抗血管新生および抗腫瘍活性を示した、KIT、PDGFR、VEGFR-1、VEGFR-2、VEGFR-3、FLT3、RETを阻害する経口マルチキナーゼ阻害薬である。スニチニブとイマチニブは共にKITおよびPDGFRのATP結合ドメイン内で結合するが、異なる結合特性と親和性を有しており、更にスニチニブは腫瘍関連の血管新生に重要なVEGFRを阻害する(イマチニブにはない特性)。このような相違によりスニチニブはイマチニブ耐性のGIST症例に臨床的有用性をもたらす可能性があると推察された。
スニチニブの第1/2相試験ではイマチニブ耐性GISTに対して奏効割合は低かったものの、臨床的に意義のある病勢制御割合を示し、有望な抗腫瘍効果を示した。イマチニブ不応後に有効な薬剤が承認されていないこともあり、イマチニブ不応/不耐のGIST患者に対するに対するスニチニブの有効性と安全性を評価する目的で本試験が計画された。
KITおよびPDGFRのキナーゼ活性を選択的に阻害するイマチニブは進行GISTの有効性を大幅に改善した。しかし進行GISTの5%はイマチニブに対する一次耐性を、また14%は早期耐性を示し、獲得耐性は治療開始後約2年(中央値)に出現する。しかし、イマチニブ不応となったGISTに対する有効な治療薬はなく、新たな治療薬の開発はアンメットメディカルニーズであった。
スニチニブはin vitroおよびin vivo腫瘍モデルで抗血管新生および抗腫瘍活性を示した、KIT、PDGFR、VEGFR-1、VEGFR-2、VEGFR-3、FLT3、RETを阻害する経口マルチキナーゼ阻害薬である。スニチニブとイマチニブは共にKITおよびPDGFRのATP結合ドメイン内で結合するが、異なる結合特性と親和性を有しており、更にスニチニブは腫瘍関連の血管新生に重要なVEGFRを阻害する(イマチニブにはない特性)。このような相違によりスニチニブはイマチニブ耐性のGIST症例に臨床的有用性をもたらす可能性があると推察された。
スニチニブの第1/2相試験ではイマチニブ耐性GISTに対して奏効割合は低かったものの、臨床的に意義のある病勢制御割合を示し、有望な抗腫瘍効果を示した。イマチニブ不応後に有効な薬剤が承認されていないこともあり、イマチニブ不応/不耐のGIST患者に対するに対するスニチニブの有効性と安全性を評価する目的で本試験が計画された。
シェーマ
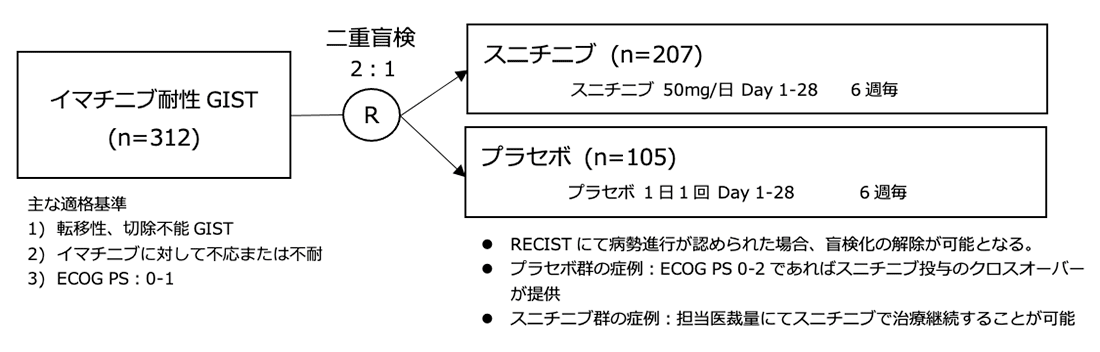
統計学的事項
主要評価項目:腫瘍増悪までの期間*(Time to Tumor Progression, 中央判定)
2002年に進行GISTに対する唯一、有効な治療であるイマチニブが臨床導入されていたが、本試験の計画時点(2002-2003年)でイマチニブ不応後の予想される臨床経過に関するデータはほとんど発表されていなかった。よって、進行GISTに対してイマチニブを定期的に使用している世界中の25名の専門家を対象に非公式の調査を実施したところ、イマチニブ不応後の腫瘍増悪までの期間は一般的に4ヶ月未満であると報告された。スニチニブの投与により、腫瘍増悪までの期間が4ヶ月から6ヶ月に改善(HR 0.67)することは臨床的に意味があると判断され、両側α=0.05、検出力を90%とすると281イベントが必要であると推定された。最小限の観察期間で281イベントを観察するにはスニチニブ群 238例、プラセボ群 119例、合計 357例の登録が必要と見込まれた。
また、141イベント、211イベント発生時点で中間解析を実施する旨、プロトコールで定義された。
*RECISTで定義された増悪のみをイベントとする
試験結果:
- 2003年12月から2005年1月の間に11ヶ国56施設より312例が登録され、無作為割り付けされた(スニチニブ群 n=207、プラセボ群 n=105)。
- 本試験では149イベント発生時に実施した初回の中間解析において、プラセボ群と比較し、スニチニブ群で有意に腫瘍増悪までの期間を延長したため、安全性モニタリング委員会の推奨に基づき、盲検化は解除され、プラセボ群はスニチニブへのクロスオーバーが許容された。データカットオフは中間解析の時点となった。
1. 患者背景
| N (%) | スニチニブ群 (N=207) | プラセボ群 (N=105) | |
|---|---|---|---|
| 年齢 (歳) | 中央値 (範囲) | 58.0 (23-84) | 55.0 (23-81) |
| 性別 | 男性 女性 |
132 (63.8) 75 (36.2) |
64 (61.0) 41 (39.0) |
| ECOG PS | 0 1 2* |
92 (44.4) 113 (54.6) 2 (1.0) |
48 (45.7) 55 (52.4) 2 (1.9) |
| 組織型 | 紡錘形細胞 混合型 (紡錘形+上皮様) 上皮様細胞 その他 不明 |
125 (60.4) 33 (15.9) 17 (8.2) 31 (15.0) 1 (0.5) |
74 (70.5) 13 (12.4) 7 (6.7) 10 (9.5) 1 (1.0) |
| 登録時の腫瘍径 (mm) | 中央値 (範囲) | 233 (26-722) | 239 (29-749) |
| イマチニブの最大投与量 (mg) | 中央値 (範囲) | 800 (300-1600) | 800 (400-1600) |
| イマチニブの1日投与量 (mg) | 中央値 | 503 | 485 |
| イマチニブの累積投与量 (mg) | 中央値 | 367,400 | 376,400 |
| イマチニブの投与期間 (週) | 中央値 (範囲) | 105.3 (0.3-205.1) | 106.9 (11.4-187.7) |
| イマチニブの治療経過 | 6ヶ月以内に増悪 6ヶ月以降に増悪 不耐 |
36 (17.4) 162 (78.3) 9 (4.3) |
17 (16.2) 84 (80.0) 4 (3.8) |
| イマチニブの最良効果 | CR PR SD PD 該当なし/不明 |
6 (2.9) 51 (24.6) 87 (42.0) 58 (28.0) 5 (2.4) |
1 (1.0) 36 (34.3) 36 (34.3) 30 (28.6) 2 (1.9) |
*より早期のスクリーニング時点ではECOG PS 2未満であり、適格と判断
- スニチニブ群とプラセボ群との間に患者背景における大きな隔たりは認めなかった。
- 主な転移臓器は肝、腹膜、腸間膜であった。
2. 腫瘍増悪までの期間 (主要評価項目:Time to Tumor Progression, 中央判定)
| N | 中央値 | (95%信頼区間) | |
|---|---|---|---|
| スニチニブ群 | 207 | 27.3週 | (16.0-32.1) |
| プラセボ群 | 105 | 6.4週 | (4.4-10.0) |
HR 0.33 (95%信頼区間 0.23-0.47), p<0.0001
- 年齢、体重、性別、人種、初回診断時からの時間、ECOG PS、McGill疼痛質問票スコア、イマチニブ投与期間/最大投与量、国で行われたサブグループ解析では全てのサブグループでHR 0.5未満であった。
3. 投与状況 (盲検化解除時点)
| スニチニブ群 | プラセボ群 | |
|---|---|---|
| 治療サイクル数 中央値 (範囲) | 2サイクル (0-9) | 1サイクル (0-6) |
| 内服日数 中央値 (範囲) | 56.0日 (1-236) | 29.5日 (2-168) |
- 減量を要した症例:スニチニブ群 23例(11%)、プラセボ群 0例
- 休薬を要した症例:スニチニブ群 57例(28%)、プラセボ群 20例(20%)
- データカットオフの段階でスニチニブ群 134例(65%)、プラセボ群 34例(32%)が試験治療を継続していた。試験治療を中止した最も多い理由は病勢進行(スニチニブ n=51、プラセボ n=65)と有害事象(スニチニブ n=15、プラセボ n=3)だった。
- スニチニブ群 19例(9%)、プラセボ群 59例(56%)は画像評価による病勢進行確認後、非盲検スニチニブ投与に移行した。
4. 無増悪生存期間
| N | 中央値 | (95%信頼区間) | 26週(6ヶ月)無増悪生存割合 | |
|---|---|---|---|---|
| スニチニブ群 | 207 | 24.1週 | (11.1-28.3) | 16% |
| プラセボ群 | 105 | 6.0週 | (4.4-9.9) | 1% |
HR 0.33 (95%信頼区間 0.24-0.47), p<0.0001
5. 全生存期間
- スニチニブ群の多くの症例は中間解析時点で生存しており、中央値を算出することはできなかった。
- HR 0.49 (95%信頼区間 0.29-0.83), p=0.007
6. 奏効割合
| スニチニブ群 (n=207) | プラセボ群 (n=105) | p値 | |
|---|---|---|---|
| CR PR SD PD |
0 14 (7%) 120 (58%) 39 (19%) |
0 0 50 (48%) 39 (37%) |
― |
| 奏効割合 (95%信頼区間) |
14 (7%) (3.7-11.1) |
0 (0%) ― |
0.006 |
- イマチニブ不応後の症例の奏効割合(10/198, 5.1%)も、全集団の奏効割合と同様であった。
- プラセボ群からスニチニブ投与にクロスオーバーした59例の内、6例でPRを得た(奏効割合 10.2%、95%信頼区間 3.8-20.8)。他、4例はクロスオーバー後、少なくとも26週はSDを維持した。
- スニチニブ群の奏効までの期間中央値:10.4週(95%信頼区間 9.7-16.1)
- スニチニブ群の奏効例(14例)中、中間解析事典で増悪を示したのは3例のみであり、奏効期間を算出することはできなかった。この増悪を示した3例の奏効期間は15.9-29.9週であった。
- 本試験にイマチニブ不耐後に登録された13例の内、スニチニブ群には9例が無作為割付されたが、9例中4例がPRを得て、解析時に病勢進行を来したのは1例のみであった。症例数は少ないものの、奏効割合はイマチニブ不応後の症例よりイマチニブ不耐後の症例で良好な傾向を示した。
7. 有害事象 (CTCAE v3.0)
| スニチニブ群 (n=202) | プラセボ群 (n=102) | |||||
|---|---|---|---|---|---|---|
| Grade 1/2 | Grade 3 | Grade 4 | Grade 1/2 | Grade 3 | Grade 4 | |
| 非血液学的有害事象 疲労 下痢 皮膚変色 悪心 食欲不振 味覚変化 口内炎 嘔吐 手足の皮膚反応 皮疹 無力症 粘膜炎 消化不良 高血圧 鼻出血 髪色の変化 口内感想 舌の痛み |
58 (29) 52 (26) 50 (25) 47 (23) 38 (19) 36 (18) 30 (15) 30 (15) 19 (9) 24 (12) 18 (9) 24 (12) 22 (11) 15 (8) 14 (7) 14 (7) 13 (6) 11 (6) |
10 (5) 7 (3) 0 (0) 1 (1) 0 (0) 0 (0) 1 (1) 1 (1) 9 (4) 2 (1) 6 (3) 0 (0) 1 (1) 6 (3) 0 (0) 0 (0) 0 (0) 0 (0) |
0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) |
20 (20) 8 (8) 6 (6) 10 (10) 5 (5) 2 (2) 2 (2) 5 (5) 2 (2) 5 (5) 2 (2) 0 (0) 1 (1) 4 (4) 0 (0) 2 (2) 1 (1) 0 (0) |
2 (2) 0 (0) 0 (0) 1 (1) 1 (1) 0 (0) 0 (0) 1 (1) 0 (0) 0 (0) 2 (2) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) |
0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) 0 (0) |
| 血液学的有害事象 貧血 白血球減少 好中球数減少 リンパ球数減少 好中球数減少 |
117 (58) 104 (52) 86 (43) 80 (40) 72 (36) |
7 (4) 7 (4) 17 (8) 18 (9) 8 (4) |
0 (0) 0 (0) 3 (2) 1 (1) 1 (1) |
59 (58) 5 (5) 4 (4) 31 (30) 4 (4) |
2 (2) 0 (0) 0 (0) 2 (2) 0 (0) |
0 (0) 0 (0) 0 (0) 1 (1) 0 (0) |
- 重篤な治療関連有害事象:スニチニブ群 40例(20%)、プラセボ群 5例(5%)
- 有害事象による治療中止:スニチニブ群 19例(9%)、プラセボ群 8例(8%)
- スニチニブ群の8例(4%)で甲状腺機能低下症を発症し、その内の1例はGrade 4だった。
- イマチニブ不耐だった症例もイマチニブ投与中に経験した有害事象の再燃なく、スニチニブは認容可能であった。
結語
イマチニブ不応/不耐後の進行消化管間質腫瘍症例において、スニチニブはプラセボと比較して、病勢制御及び生存期間を含む臨床的有用性を有意に改善した。また安全性は許容範囲内であった。
執筆:神奈川県立がんセンター 消化器内科 医長 古田 光寛 先生
監修:近畿大学医学部 内科学教室 腫瘍内科部門 医学部講師 川上 尚人 先生
監修:近畿大学医学部 内科学教室 腫瘍内科部門 医学部講師 川上 尚人 先生